

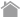
Human class I HLA-E Tetramer
HLA-E Tetramer can be used for
direct detection of antigen specific NK cells and T cells.
 Like classical MHC molecules, the non-classical MHC class I molecule HLA-E is deeply involved in self/nonself recognition and its associated immune response. Unlike classical MHC molecules, however, non-classical MHC molecules exhibit little polymorphism, and in the case of HLA-E, Japanese people mainly fall into the following two categories: HLA-E*01:01 or HLA-E*01:03.
Like classical MHC molecules, the non-classical MHC class I molecule HLA-E is deeply involved in self/nonself recognition and its associated immune response. Unlike classical MHC molecules, however, non-classical MHC molecules exhibit little polymorphism, and in the case of HLA-E, Japanese people mainly fall into the following two categories: HLA-E*01:01 or HLA-E*01:03.
HLA-E inhibits the cellular immune response by binding to the CD94/NKG2 receptor expressed on NK and CD8+ T cells1). In addition, it is known that HLA-E promotes activation by binding to the T cell receptor (TCR) expressed on CD8+ T cells2).
It has been reported that HLA-E is expressed in melanoma, osteosarcoma, and other cancer cells, and that the tumor cells escape from NK and CD8+ T cells immune surveillance by binding to inhibitory CD94/NKG2A receptor via HLA-E3,4,5). HLA-E functions have also been implicated in infectious diseases2,6,7), and the role of HLA-E in these diseases is currently under scrutiny.
HLA-E*01:03 HLA-A leader3-11 Tetramer-VMAPRTLVL-PE
It is known that HLA-E preferably binds to a peptide derived from HLA-A, -B, -C, and -G leader sequences (3-11 aa)1,8). Under physiological conditions, HLA-E bound to such an HLA class I leader sequence binds to the CD94/NKG2A receptor expressed on NK and CD8+ T cells, resulting in the transduction of an inhibitory signal that suppresses cytotoxicity. It has been reported that cytomegalovirus (CMV) escapes from NK cell immune surveillance by expressing the UL40 protein, which has a sequence mimicking an HLA class I leader sequence9). It has also been reported, however, that CD8+ T cells recognize HLA-E bound to an HLA class I leader/CMV UL40 peptide sequence and become involved in defense against CMV infection2).
Example 1
Staining Examples of HLA-E HLA-A leader3-11 Tetramer and HLA-E Negative Tetramer
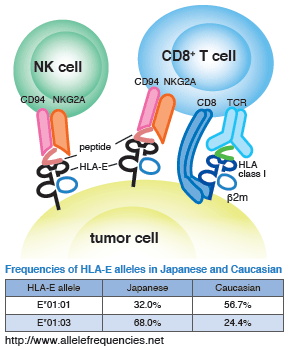
 Staining of PBMC isolated from healthy individuals was carried out using HLA-E*01:03 HLA-A leader3-11 Tetramer-VMAPRTLVL-PE (Code No. TS-ME01-1) and HLA-E*01:03 Negative Tetramer-VMAPKTLVL-PE (Code No. TS-ME02-1). The results showed that when the HLA-E*01:03 HLA-A leader3-11 Tetramer was used, both tetramer-positive CD8− and CD8+ cells were detected.
Staining of PBMC isolated from healthy individuals was carried out using HLA-E*01:03 HLA-A leader3-11 Tetramer-VMAPRTLVL-PE (Code No. TS-ME01-1) and HLA-E*01:03 Negative Tetramer-VMAPKTLVL-PE (Code No. TS-ME02-1). The results showed that when the HLA-E*01:03 HLA-A leader3-11 Tetramer was used, both tetramer-positive CD8− and CD8+ cells were detected.
The numbers in the plots represent the ratio of tetramer-positive cells (%) to total lymphocytes in each panel.
Example 2
HLA-E Tetramer Binding Inhibition by CD94 Antibody
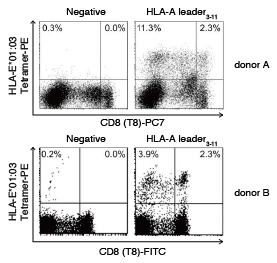
 After PBMCs isolated from healthy individuals were preincubated with CD94 antibody (clone DX22), they were stained with HLA-E*01:03 HLA-A leader3-11 Tetramer-PE1). The results showed that the CD94 antibody inhibited binding of the HLA-E Tetramer to the CD94/NKG2 receptor in both the CD8− and CD8+ cells. In addition, the findings also indicated that during HLA-E Tetramer binding inhibition by the CD94 antibody, the observed population of tetramer-positive CD8+ cells recognized the HLA-E Tetramer via receptors other than CD94/NKG2.
After PBMCs isolated from healthy individuals were preincubated with CD94 antibody (clone DX22), they were stained with HLA-E*01:03 HLA-A leader3-11 Tetramer-PE1). The results showed that the CD94 antibody inhibited binding of the HLA-E Tetramer to the CD94/NKG2 receptor in both the CD8− and CD8+ cells. In addition, the findings also indicated that during HLA-E Tetramer binding inhibition by the CD94 antibody, the observed population of tetramer-positive CD8+ cells recognized the HLA-E Tetramer via receptors other than CD94/NKG2.
The numbers at the top right of each panel represent the ratio of tetramer-positive cells (%) to total lymphocytes, and the numbers in parentheses indicate the ratio of tetramer-positive cells (%) to total CD8+ cells.
Example 3
HLA-E Tetramer Binding Inhibition by CD3 Antibody
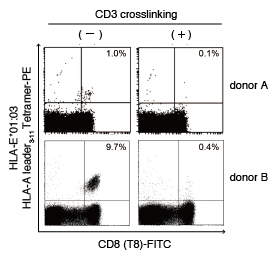 After PBMCs isolated from healthy individuals were preincubated with CD3 antibody, they were rinsed and incubated with anti-mouse IgG secondary antibody 10). Cells were rinsed again, incubated with CD94 antibody (clone DX22), and stained with HLA-E*01:03 HLA-A leader3-11 Tetramer-PE. The results showed that in the PBMCs from donor A and B, binding of the HLA-E Tetramer to the TCR/CD3 complex was inhibited by the CD3 antibody/anti-mouse IgG secondary antibody treatment.
After PBMCs isolated from healthy individuals were preincubated with CD3 antibody, they were rinsed and incubated with anti-mouse IgG secondary antibody 10). Cells were rinsed again, incubated with CD94 antibody (clone DX22), and stained with HLA-E*01:03 HLA-A leader3-11 Tetramer-PE. The results showed that in the PBMCs from donor A and B, binding of the HLA-E Tetramer to the TCR/CD3 complex was inhibited by the CD3 antibody/anti-mouse IgG secondary antibody treatment.
The numbers in the plots represent the ratio of tetramer-positive cells (%) to total CD8+ cells.
Type of NKG2 molecules that form heterodimers with CD94 molecules
| NKG2A | inhibitory |
| NKG2B | inhibitory |
| NKG2C | activating |
References
- Braud VM, et al. Nature 391: 795-799 (1998) PMID: 9486650
- Pietra G, et al. PNAS 100: 10896-10901 (2003) PMID: 12960383
- Monaco EL, et al. Neoplasia 13: 822-830 (2011) PMID: 21969815
- Speiser DE, et al. J Exp Med 190: 775-782 (1999) PMID: 10499916
- Derré L, et al. J Immunol 177: 3100-3107 (2006) PMID: 16920947
- Heinzel AS, et al. J Exp Med 196: 1473-1481 (2002) PMID: 12461082
- Salerno-Gonçalves R, et al. J Immunol 173: 5852-5862 (2004) PMID: 15494539
- Miller JD, et al. J Immunol 171: 1369-1375 (2003) PMID: 12874227
- Tomasec P, et al. Science 287: 1031-1033 (2000) PMID: 10669413
- Weder P, et al. Results in Immunology 2: 88-96 (2012) PMID: 24371571