

Articles from key opinion leaders
Autophagy research: Current status and future perspectives
 Dr. Noboru Mizushima
Dr. Noboru Mizushima
of the University of Tokyo
1. What is autophagy?
The lysosome is a cellular organelle whose main function is degradation. It is primarily known as the place where extracellular materials and plasma membrane proteins internalized by endocytosis are degraded. But, it can certainly degrade intracellular components as well (Figure 1). Autophagy is a “cellular function in which the cell degrades its own components in the lysosome.” Although often mistaken for a type of cell death, autophagy is a degradative process that mostly protects the cell from cell death.
Autophagy is broadly classified into “macroautophagy,” “microautophagy,” and “chaperone-mediated autophagy” (Figure 1). Among them, macroautophagy has the largest degradative capacity and has been extensively studied in many species from yeast to animals and plants. In contrast, microautophagy and chaperone-mediated autophagy have been studied primarily in yeast and mammals, respectively, and their occurrence and molecular mechanisms are not fully understood (microautophagy might be the same process as the formation of multivesicular bodies in mammals). For this reason, macroautophagy is commonly referred to by the term autophagy, which will be used hereafter in this article.
Many of the molecules involved in autophagy were identified in genetic studies of the budding yeast in early 1990s by Dr. Yoshinori Ohsumi (currently of the Tokyo Institute of Technology) and his colleagues [1]. The ATG1 through ATG41 genes have been known as of August 2016. Most of these genes are required for selective autophagy, which targets specific substrates for degradation. For example, ATG30 is required only for autophagy of peroxisomes (pexophagy), and ATG32 is required only for autophagy of mitochondria (mitophagy). In contrast, the 15 genes known as the “core ATG genes” (ATG1 – 10, 12 – 14, 16, 18) are required for all types of autophagy, including the non-selective “ordinary autophagy” that is induced during starvation. These genes are highly conserved in other organisms including mammals. The detailed functions of these genes have been reviewed elsewhere [2, 3].
Figure 1. Three types of autophagy
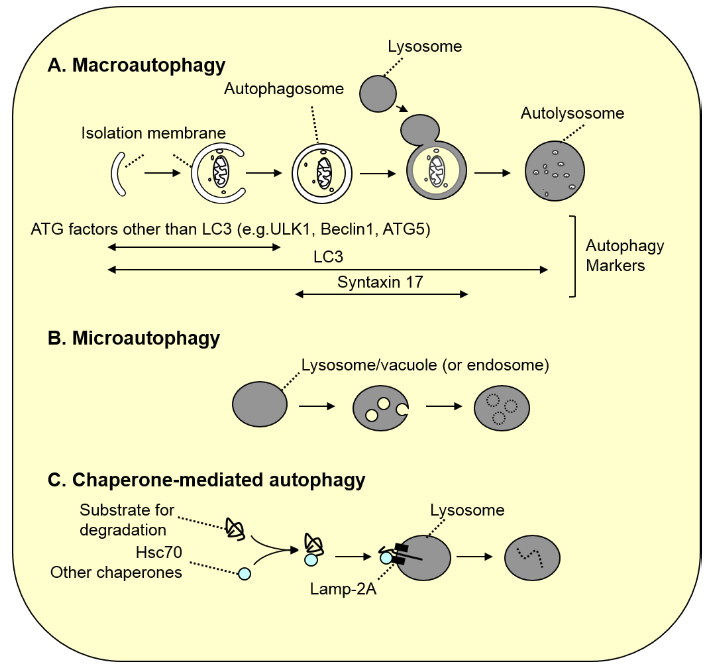
A. Macroautophagy. As autophagosomes (approximately 1 µm in diameter) are formed, a portion of the cytoplasm is enclosed. Subsequently, the autophagosomes fuse with the lysosome, resulting in degradation of the inner autophagosomal membrane along with its contents. Because LC3 family proteins are localized to most membranes formed in this process, LC3 is used as a general marker for autophagy-associated membranes (in autolysosomes, LC3 on the inner membrane is degraded, and LC3 on the outer membrane is gradually detached from the membrane). In contrast, most ATG factors other than LC3 are localized only to the isolation membrane prior to closure, and syntaxin 17, which is necessary for fusion with the lysosome, is localized to autophagosomes after closure. Thus, these molecules serve as specific markers for these stages.
B. Microautophagy. Lysosomal or vacuolar membranes invaginate and detach, thereby engulfing the cytoplasmic components. In yeast, peroxisomes and a portion of the nucleus are degraded in this process (in microautophagy of peroxisomes, vacuolar membrane is not sufficient for the engulfment, and de novo membrane synthesis is required). In mammals, late endosomal membranes undergo similar membrane dynamics known as “endosomal microautophagy” to degrade cytoplasmic components.
C. Chaperone-mediated autophagy. Cytoplasmic proteins with the pentapeptide “KFERQ motif” are recognized by Hsc70 and other co-chaperones, and directly transported into the lumen of the lysosome via binding to the LAMP2A receptor on the lysosome.
2. Current status of autophagy research
The discovery of yeast ATG genes opened an entirely new chapter of autophagy research, in which three main features have to be considered together.
First, functional analysis of the yeast Atg molecules was conducted using powerful yeast genetics as the main tool. Studies using genetic, biochemical, morphological, and structural biology approaches broadly advanced our understanding of various aspects of autophagy, such as the function of individual Atg proteins and complexes, their genetic hierarchy, the mechanism of regulation of their activities, and the mechanism of substrate recognition. In yeast, the autophagosome is formed from the “pre-autophagosomal structure (PAS),” which typically exists as a single entity near the vacuole. Through details of autophagosome formation have been actively studied, many questions remain unanswered, including the origin of the autophagosomal membrane, composition of proteins and lipids in the membrane, mechanism of autophagosome membrane closure, and mechanism of fusion with the vacuole.
Second, ATG homologues have been studied in species other than yeast. Even before genome sequences were completed, it was clear that many species had ATG homologues, including mammals and plants. Although red algae and some protozoan species lack some or all ATG homologues [4], ATG genes are conserved in nearly all eukaryotes. In addition, autophagy factors that are absent in yeast, such as VMP1, EPD5, and Ei24, were discovered in higher animals. These discoveries complement those made by yeast studies on the molecular mechanism of autophagy, and at the same time made important contributions to the development of markers essential for autophagy research. A good example is the autophagosome marker LC3B, which was discovered by Dr. Tamotsu Yoshimori (currently of Osaka University), and is most widely used today (Figure 1) [5, 6]. In many organisms other than the budding yeast, autophagosomes are simultaneously formed at multiple locations in the cell. These locations are closely associated at least with the endoplasmic reticulum.
Third, phenotypes of atg null mutants have been investigated in various species using the reverse genetics approach. Numerous tissue-specific ATG knockout mice have been generated in addition to simple knockout mice. For example, the ATG5flox mice, generated in the author’s laboratory, alone have been shared with more than 400 laboratories. Presumably, this strain has been crossed with all publicly available Cre-expressing mice. As a result, it has become clear that autophagic degradation essentially plays two main roles [7]. One is to supply degradation products, which is important in maintaining the intracellular amino acid pool during starvation and early embryogenesis, and for self-antigen presentation. The first evidence of this role was the isolation of yeast autophagy mutants that had lost resistance to starvation [1]. This role of autophagy is believed to be common to all organisms. The second role is intracellular quality control via substrate-selective as well as non-selective bulk degradation. Substrates of selective autophagy include LC3-binding proteins such as p62/SQSTM1, damaged organelles such as depolarized mitochondria, and intracellular pathogens (in some cases, damaged endosomes surrounding bacteria). As demonstrated by Dr. Masaaki Komatsu (currently of Niigata University), it is especially important to maintain p62/SQSTM1 at a low level in order to prevent accumulation of protein aggregates and uncontrolled activation of the oxidative stress-responsive Nrf2 pathway. Insufficient quality control is generally associated with cellular degeneration in the nervous system and the liver, acceleration of age-related changes, tumor formation, and exacerbation of infection in mice. These quality control functions have a greater impact on long-lived cells and little impact on cells (such as budding yeast) whose doubling time is much shorter than the average half-life of proteins. Similar physiological studies in many non-mouse models have been reported, but are not included in this article due to limited space.
3. Autophagy and human disease
Basic research on autophagy has dramatically expanded in the past 10 years. In contrast, our understanding of the relevance of autophagy in medicine has lagged far behind. Only recently, association of autophagy with human disease has begun to emerge through studies in human genetics.
In 2007, ATG16L1 was identified as a risk allele associated with the inflammatory bowel disease, Crohn’s disease [8, 9]. This allele does not encode a null mutation, but has a threonine-to-alanine substitution at position 300 (T300A), which is located close to the center of ATG16L1. Healthy individuals often carry this allele, and the odds ratio for Crohn’s disease is about 2-fold in patients with the homozygous mutation. The C-terminal domain that includes the mutated region is not present in yeast Atg16, and the function of this domain remains largely unknown in mammals. There are conflicting reports about the T300A mutation; some claimed that the mutation affected the activity of autophagy, and others reported the opposite. Regardless, given that the high-frequency mutation was identified through statistical analyses of multiple populations, it would be very challenging to define the significance of the mutation in tissue culture cells or experimental animals.
Dr. Richard Youle and his colleagues in the United States reported in 2008 that a causative gene for familial Parkinson’s disease, Parkin/PARK2, was involved in autophagic degradation of depolarized mitochondria (also called mitophagy) [10]. Parkin is a ubiquitin ligase localized to depolarized/damaged mitochondria. The localization process requires another factor associated with familial Parkinson’s disease, PINK1/PARK6, and phosphorylated ubiquitin [11]. Based on these findings, Parkin-associated Parkinson’s disease and PINK1-associated Parkinson’s disease are thought to be caused by impaired mitophagy, resulting in accumulation of damaged mitochondria that should have been degraded. However, Parkin has other functions unrelated to mitophagy, such as induction of outer membrane protein degradation while maintaining intact mitochondria [12]. Additional in vivo experiments are necessary to demonstrate that impaired mitophagy itself is the cause of Parkinson’s disease.
Since 2012, mutations in autophagy-related genes have been identified one after another through exome analyses of patients’ families. Dr. Hayflick’s group in the United States and Drs. Naomichi Matsumoto and Hirotomo Saitsu (currently of Yokohama City University) independently discovered mutations in WDR45/WIPI4 gene as a cause for SENDA (static encephalopathy of childhood with neurodegeneration in adulthood) [13, 14]. WIPI4 is the human homologue of yeast ATG18 or ATG21 (and there are four WIPI proteins 1 – 4). Moreover, this is the first report of human disease associated with mutations in core ATG genes. WDR45 gene is located on the X chromosome, and most cases occur in women with mosaicism. SENDA (also known as BPAN) is a neurodegenerative disease characterized by iron accumulation in the basal ganglia in the brain. Patients present with non-progressive intellectual and motor deficits in childhood, and their Parkinson-like symptoms rapidly progress after 20 – 30 years of age. We demonstrated that patients’ lymphoblasts had reduced autophagic activity [14]. However, the exact function of WIPI4 remains unclear. Among the WIPI family proteins, WIPI2 has the most important role in typical culture cells such as HeLa cells. In contrast, p62, which is a substrate of selective autophagy, is found to accumulate in the nerves of the recently generated WIPI4 knockout mice, suggesting the possibility that WIPI4 plays an important role in the nervous system [15]. As for mutations in other core ATG genes, an ATG5 mutation has been reported in patients with congenital ataxia with mental retardation [16]. This mutation results in partially reduced activity of autophagy due to impaired ATG5-ATG12 conjugation. Mutations in other genes have also been identified through exome analyses. For example, mutations in a gene involved in autolysosome degradation, EPG5, were identified in patients with Vici syndrome (characterized by agenesis of the corpus callosum, cataracts, cardiomyopathy, immunodeficiency, and hypopigmentation) [17]; a mutation in TECPR2 gene, encoding an LC3-binding protein, was identified in patients with hereditary spastic paraparesis [18]; and mutations in lysosomal PI(3,5,)P2-binding protein SNX14 were identified in patients with cerebellar atrophy [19]. These mutations cause reduced activity of autophagy as well. However, it is likely that these effects are primarily caused by lysosomal abnormalities rather than the autophagy pathway per se. See another review for details [20].
4. Therapeutic approaches targeting autophagy
To date, autophagy is known as the direct cause of human disease in only a limited number of cases. Nevertheless, efforts to target autophagy as a therapeutic strategy have already begun. In fact, a drug chloroquine (or hydroxychloroquine) is being tested in clinical trials for malignant tumors [20]. Chloroquine inhibits lysosomal function. This drug is not necessarily specific for autophagy. A multicenter clinical trial is currently underway with the University of Pennsylvania in the United States as the coordinating center. As of August 2016, 27 studies with hydroxychloroquine and 8 studies with chloroquine have been registered on the NIH’s website ClinicalTrials.gov (https://clinicaltrials.gov/ct2/home) as Phase 1 or 2 trials. Most of the studies are combination trials. Detailed outcomes of some of the trials have been reported in 6 articles published in the August 2014 issue of Autophagy. According to the reports, treatments were effective in some patients. Several theories have been proposed to explain why inhibition of autophagy can be effective for cancer treatment. The therapeutic effects could be attributed to inhibition of a broad range of autophagic functions, such as cellular remodeling and a quality control mechanism, in addition to inhibition of amino acid production [22, 23]. If a subset of cancers could be identified as highly dependent on autophagy, treatment could be more effective. On the other hand, the anticancer effects of chloroquine may be independent of inhibition of autophagy [24]. Thus, effects of autophagy inhibitors with higher specificity should be tested in the future.
Although neurodegenerative disease could possibly be effectively treated by targeting autophagy, large-scale clinical trials are yet to be conducted. Many neurodegenerative diseases are caused, in part, by accumulation of abnormal proteins in the cell. Therefore, efforts are being made to remove abnormal or denatured proteins that are harmful to the cell, by enhancing the intracellular cleansing effect of autophagy. In experiments using neurodegenerative disease models (such as polyglutamine disease in mouse and Drosophila), the mTORC1 inhibitor rapamycin and its derivatives were effective in reducing the symptoms. However, mTORC1 inhibitors are not suitable for human use because of strong side effects. Hence, mTORC1-independent activators of autophagy have been sought. One such drug is the antiepileptic medicine carbamazepine. A report showed that carbamazepine was effective in an animal model of alpha1-antitrypsin deficiency, in which liver damage was caused by accumulation of the mutant protein in the endoplasmic reticulum of hepatocytes [25]. Although it is unclear whether the treatment effects were truly mediated by activation of autophagy, future developments are expected in this area.
5. Future tasks and perspectives
Much has been discovered about the mechanism and physiological significance of autophagy. Although many important issues remain to be addressed, continued progress toward elucidation is expected in the future. While at the same time, autophagy-associated factors have been found to be also involved in physiological pathways other than autophagy. The findings in this area include the “LC3-associated phagocytosis (LAP)” pathway in which autophagy factors (excluding the ULK complex) facilitate the maturation of phagosomes, and a different pathway in which autophagosomes or related structures are used in non-classical secretion [26, 27]. The physiological significance of these processes is currently under investigation, and is thought to be closely associated with immune processes, such as phagocytosis of dead cells, autoimmune disease, and secretion of cytokines.
There is much room for improvement in methods for monitoring autophagy in vivo. Even in basic research using tissue culture cells, current methods of autophagy measurement are still complicated and not fully satisfactory. For instance, an increase in the number of autophagosomes and the LC3-II form (the membrane-bound form of LC3) does not unconditionally indicate activation of autophagy. Instead, it could indicate a blockage at a later step of autophagy, such as inhibition of lysosomes [6]. In fact, in previously conducted screening of compounds, increased number of LC3 puncta or LC3-II levels had mainly resulted from blockage at a later stage of the process and not activation of autophagy. Like chloroquine, many weak base compounds have such characteristics. This issue has become well recognized recently, and a more appropriate “flux assay” has been commonly used in parallel. However, it is difficult or impossible to perform a flux assay using fixed samples, and this difficulty is a major impediment in histopathological analyses. Further, because these assays can be done only with dissected tissue samples, it is virtually impossible to measure the activity of autophagy in living human beings at this time. An indirect method to assess the activity of autophagy needs to be developed, even if it might be imperfect. With such a method, it may become possible to identify human disease with partially impaired autophagy. Currently, there are very few ways to detect the involvement of autophagy, other than identifying mutations in autophagy-associated genes.
Lastly, regarding autophagy as the target for drug discovery, there will be plenty of possibilities in this area. A frequently expressed concern is that activating autophagy may have side effects. But, we view this issue optimistically. Notably, knocking out the relatively low steady-state level of autophagy can cause marked accumulation of abnormal proteins. This means that the low steady-state level of autophagy activity has a sufficient intracellular cleansing effect. Therefore, a slight increase in activation can be expected to have significant effects. Also, since there must be a sophisticated feedback mechanism between intracellular degradation and synthesis, activation of degradation is likely to be offset by activation of synthesis. If turnover (and not degradation alone) is increased, toxicity may not be very high. Hence, it would be more important to develop drugs that exclusively activate the autophagy pathway.
References
1. Tsukada, M. and Y. Ohsumi, FEBS Lett., 1993. 333:169-174.
2. Nakatogawa, H., et al., Nat. Rev. Mol. Cell Biol., 2009. 10:458-67.
3. Mizushima, N., T. Yoshimori, and Y. Ohsumi, Annu. Rev. Cell Dev. Biol., 2011. 27:107-132.
4. Shemi, A., S. Ben-Dor, and A. Vardi, Autophagy, 2015. 11:701-15.
5. Kabeya, Y., et al., EMBO J., 2000. 19:5720-5728.
6. Mizushima, N., T. Yoshimori, and B. Levine, Cell, 2010. 140:313-26.
7. Mizushima, N. and M. Komatsu, Cell, 2011. 147:728-41.
8. Hampe, J., et al., Nat. Genet., 2007. 39:207-11.
9. Rioux, J.D., et al., Nat. Genet., 2007. 39:596-604.
10. Narendra, D., et al., J Cell Biol, 2008. 183:795-803.
11. Durcan, T.M. and E.A. Fon, Genes Dev., 2015. 29:989-999.
12. Scarffe, L.A., et al., Trends Neurosci., 2014. 37:315-24.
13. Haack, T.B., et al., Am. J. Hum. Genet., 2012. 91:1144-1149.
14. Saitsu, H., et al., Nat. Genet., 2013. 45:445-449.
15. Zhao, Y.G., et al., Autophagy, 2015. 11:881-90.
16. Kim, M., et al., Elife, 2016. 5: e12245.
17. Cullup, T., et al., Nat. Genet., 2013. 45:83-7.
18. Oz-Levi, D., et al., Am. J. Hum. Genet., 2012. 91:1065-1072.
19. Akizu, N., et al., Nat. Genet., 2015. 47:528-534.
20. Jiang, P. and N. Mizushima, Cell Res., 2014. 24:69-79.
21. Amaravadi, R.K., et al., Clin. Cancer Res., 2011. 17:654-66.
22. Cheong, H., et al., Nat. Biotechnol., 2012. 30:671-678.
23. White, E., Nat. Rev. Cancer, 2012. 12:401-410.
24. Maycotte, P., et al., Autophagy, 2012. 8:200-212.
25. Hidvegi, T., et al., Science, 2010. 329:229-32.
26. Bestebroer, J., et al., Traffic, 2013. 14:1029-41.
27. Ponpuak, M., et al., Curr. Opin. Cell Biol., 2015. 35:106-116.